很多人想成为特别的人,而有一群人的愿望是成为普通人。“星星的孩子”“瓷娃娃”“牵线木偶人”“天使综合征”……这些美丽的名字,背后都有一个特殊的身份——罕见病患者。
今天是第十五个国际罕见病日,主题是“因罕而聚 明天更好”。
罕见病指新生儿发病率小于万分之一、患病率小于万分之一、患病人数小于14万的疾病。符合其中一项,即为罕见病。
虽然被称为罕见病,但“罕见病并不罕见”。罕见病80%由基因缺陷导致,50%于儿童期发病,30%于5岁前死亡,多为终生疾患。约有一半的患者为成年起病,需要全程治疗和医疗指导。目前人们已知的罕见病大概有6000-8000种,全球罕见病人数已超过3亿,中国罕见病患者人数约1680万。
“活下去,站起来,正常吃饭……”是很多罕见病患者和家庭最朴素的梦想。在这条实现梦想的路上,他们拼命扼住命运的咽喉,不放弃任何一线希望。也有无数医务工作者、科研人员、爱心人士与他们共同承担,让这一条艰辛之路有阳光雨露、少一些风雨、多一些色彩。加油,明天会更好。
“前面有了一束光,我们竭尽全力爬过去”
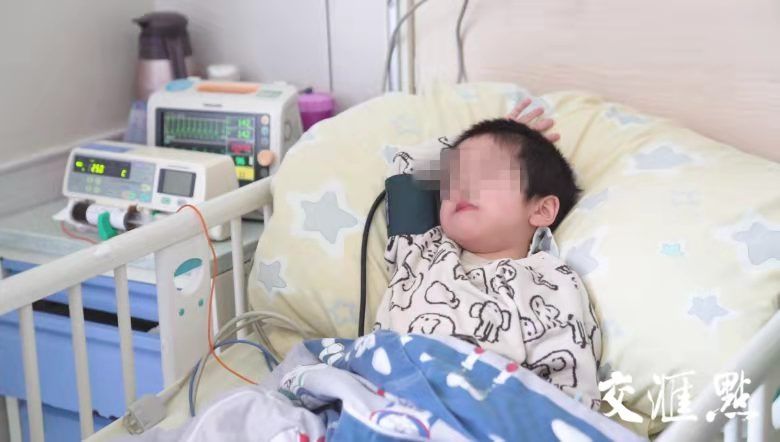
小脸蛋微微泛着红晕,眼睛澄澈而明亮。采访人员在南京市儿童医院住院部见到轩轩时,他撒着娇,把玩着橙黄色的玩具卡车,看上去就像所有其他三岁孩子一样调皮又好动。
而时不时露出的小手插满针管,一旁注射药瓶的机器闪烁着各种各样的指标,妈妈谨慎又频繁地擦拭着他难以咽下的口水……这些散落的细节提醒着人们,他病了。
2019年6月,轩轩出生在苏州一个普通双职工家庭。在他的名字中有一个“轩”字,“希望他成长为一位气宇轩昂的男子汉。”但1岁以后,轩轩的身体出现了异常:频繁感冒、呼吸困难、无法独立行走……辗转多家医院后,2021年4月,轩轩被确诊为庞贝病。
“庞贝病”全称Ⅱ型糖原贮积症,患者体内缺少一种消化酶,无法像正常人一样生长肌肉,从而出现四肢无力、肝脏肿大、呼吸困难等症状。这种病的发病率只有五万分之一,如果不及时治疗,最终将走向死亡。
文章插图
“我整个人都是懵的。”父亲刘星至今记得,确诊的那天,检测报告上显示“异常”的红色加粗字体刺得他双眼忍不住流泪。
不幸中的万幸,庞贝病有药可治,但需终身治疗,且剂量会随着体重的增长而增加。
“每次5支,每支5480元,每个月需两次,完全自费。”确诊之后,刘星决定立即给孩子用药。这也意味着,当时仅12公斤的轩轩一年就需要60多万的费用。
“治!无论如何也要坚持下去。”天价药费压得这个家庭喘不过气,刘星甚至开始联系房产中介,“如果把房子卖了,最多能撑两年,但不用药,医生评估的存活时间只有一年!”
【 让更多罕见病被“看见” 明天会更好|记录 | 佳欣】 打针太多,轩轩小手小脚上的血管都被戳破了,护士常常找不着针眼。而面对打针,轩轩已经表现得超乎一般孩子的冷静,“他习惯了。”还未体验到世间美好,却受尽人间疾苦。
刘星说,“他只要喜欢的东西,我们会尽一切努力满足他。”轩轩爱车,于是家里便摆满了小轿车、货车、吊车、工程车;轩轩爱书,尤其是打开以后可以呈现立体图案的“翻翻书”,于是刘星一套套买书;轩轩爱猫,在为数不多的可以出门晒太阳的时日,刘星会带着轩轩寻找小区的猫,阳光下,轩轩的笑脸,是世间最美好的画面。
直到2021年7月,庞贝病被纳入江苏医保基金的保障目录。刘星深深喘了一口气。虽然每个月还是会花费上万,但“只要我们努努力,就能保证孩子持续、足量的用药了!”这份保障就像是心里有了块基石,“压得人很稳,整个家庭的精神状态,气氛都好很多,笑容也越来越多。”
2月24日,刘星在病友群看到了“庞贝氏病AAV基因药物临床试验注册获审评受理”的链接,药物研发又有新进展……“我们困在一个山洞里,无数个日日夜夜,一直是黑暗的。这时候,前面有了一束光,大概率是洞口,但不管它是不是洞口,我们都要竭尽全力爬过去。”他在病友群里这样写下。
比起年幼的轩轩,同一个病房的佳欣显然承受得更多。
8年前,年仅10岁的佳欣就被确诊为庞贝病。妈妈叶女士拿着厚厚一叠检查报告,“腿都软了。”虽然有药可医,但在当时,治疗庞贝病的药还未在中国大陆上市,佳欣一家开始了漫长的等待。
为了不给孩子压力,父母对佳欣隐瞒了所有病情。但敏感细腻的佳欣还是在父母的愁容、辗转的求诊过程中有所察觉。“我知道检查报告就在那里。”医学名词看不懂,她就偷偷百度,“我查了很多次,反复地查,但关于这个病的介绍一直都是那几句话,令人非常崩溃。”
直到2021年,等待了7年的佳欣终于用上了药。直到这天,打算把实情告诉孩子的叶女士才发现,女儿已经默默承受了这么多年。“原来她都道。”
“8月23日,第一次用药,14瓶第三管刚用上时我的右腿就可以向上抬了……”
“9月6日,今天感觉不错,洗完澡之后我可以撑着站起来了!好意外!”
“9月19日,现在我独立行走的距离比第一次用药长了,也走得比之前稳了,好开心!”
……
从2021年8月23日至今,佳欣每天都坚持记录自己的变化,几十个字很短,放不下这位花季少女的彷徨、焦灼和喜悦,几十个字也很长,药物给她带来的希望可以照亮现在和未来的每一天。
“当我能够做一些治疗之前无法完成的动作时,我是无比地激动与惊喜,那种感觉就像是黑白的世界里突然多出了彩色一样。”
“世界以痛吻我,我却报之以歌”
参加升旗仪式、参与翻译社团、每天准时上网课,闲暇时间弹钢琴……这是西交利物浦大学外国语学院大一学生小兰(化名)的生活。如果不是看见她坐在轮椅上,恐怕很难想她是一名SMAⅡ 型患者。
脊髓性肌萎缩症(SMA),是一组会导致肌肉无力和萎缩的运动神经元性疾病,也是相对常见的一种“罕见病”。目前我国新生儿SMA患者每年新增约1200人,存量患者约3万人。
运动神经看上去不起眼,但对人体来说至关重要。它们起源于脊髓,控制着人体呼吸、爬、走、头颈控制以及吞咽等活动的肌肉。SMA对人周身上下的肌肉都会造成侵害,患者腿部无力的情况通常较手臂处更为严重。当参与呼吸和咳嗽的肌肉受到侵害而变得无力时,就会增加肺炎和呼吸道感染的风险。基于发病时间及所能达到的最大运动功能,SMA患者会被划分为Ⅰ、Ⅱ、Ⅲ、Ⅳ共4个类型。
小兰与大她5岁的哥哥都是SMA患者,哥哥属于SMAⅠ型,情况比她更严重。这就意味着,他们的妈妈比普通的妈妈付出更多。小兰说,从小到大,妈妈的爱和坚持让她和哥哥走得更远。
为了锻炼小兰的手指力量,在小兰7岁时,妈妈便为爱听钢琴曲的她做了一个大胆的决定——学钢琴。弹钢琴对手指力量有一定要求,小兰的手指力量本就比普通人弱,因此,必须花费异于常人的艰辛。“别人弹一遍,我可能要弹几十遍。刚开始,也想要放弃。好在,这么多年我坚持下来了,也从音乐中找到了力量与快乐。”小兰说。
上初二以前,小兰的肌肉力量可以支撑行走,但随着身体发育、课业压力增大导致运动量减少后,她逐渐需要依靠轮椅代步。“以前可以走路的时候,我就比较担心万一有一天,真的站不起来了,该怎么办?”但当这一天真正到来的时候,本以为已经做好思想准备的她还是陷入了悲伤。
天无绝人之路。2019年末。一天,爸爸方先生在《姑苏晚报》上看见了这样一篇报道:SMA精准靶向治疗药物诺西那生钠已被引进国内,并已经有苏州患儿注射。
这一刻,全家的希望被点亮。
注射诺西那生钠第一针后,要分别在第14天、第28天、第63天时连续注射3针,此后每4个月注射一针。
虽然方先生家境殷实,针剂有买有送,但近70万一针的高价还是让这个家庭喘不上气。那段时间里,白发悄悄爬上了方先生的两鬓。“两个孩子等了这么多年,如今终于有药了,就一定要治,实在不行,卖房子也行!”
2020年的小年夜当天,已经等待了十几年之久的小兰打了第一针。
从此,一切向好的方向发展——2021年7月,江苏省率先将诺西那生钠纳入地方医保;2022年1月,诺西那生钠正式被纳入国家医保。“再加上一些补贴和保险政策,一针诺西那生钠注射的费用降至1万多元。”方先生说。
“世界以痛吻我,我却报之以歌。”小兰告诉采访人员,学习生物制药曾是她的梦想。“生物医药的研发可以给很多人、很多家庭带来光明。特别我和哥哥,就是现代医学与科技的受益者。”但由于这个专业需要经常做实验,行动不便的小兰只得做罢。
2021年上半年,小兰在当时登上“热搜”的中美高层战略对话视频中发现了现场翻译张京,从此,便有了另一个理想——希望可以成为像张京那样的翻译。“如果可以的话,我也希望加入苏州残联,帮助更多需要帮助的人。”
“既然选择了远方,便只顾风雨兼程。”这是小兰最喜欢的一句话。“很庆幸我生活在这样一个家庭,成长在这样的社会。我总会鼓励自己,我有家人的陪伴、病友和那么多人的鼓励,应该活出更精彩的人生。也期待更多SMA新药的研发,让更多的和我一样的人能够打破命运的枷锁。”
在江苏省人民医院的康复门诊,采访人员见到了另一名正在进行康复治疗的SMAⅡ 型患儿浩浩(化名)。再过两个月,浩浩即将满3周岁。每天上午,妈妈刘女士都会陪着他一起在医院进行康复训练。
“或许他想在我们的怀抱中待久一点吧。”从无法接受孩子患病,到慢慢接受,再到振作起来、逐渐乐观,刘女士一直在调整心态。
“如果有一种方式,可以让更多人关注到这种罕见病,是不是可以让更多的患儿家庭少走弯路呢?如果,更多的人愿意在产前诊断时做相关的基因筛查,是不是可以避免更多罕见病患儿的出生呢?”度过了至暗时刻的张女士有了更多想法。
于是,她成为了一名“知乎”上的SMA相关话题答主,并注册海外账户,了解SMA的最新药物研发进展,也记录者儿子打针、康复过程的过程。
在“知乎”上,浩浩的妈妈这样写道:“我觉得浩浩就是缺了一条神经,就跟我天生缺心眼一样。现在起码有药可医、而且已经进入医保,一切都在朝着好的方向发展。”
“我是一个乐观的人,也希望向更多的SMA家庭传递正能量。”张女士说,“现在,我最大的心愿就是孩子可以早日站起来、走起来,健康成长,开心快乐就好。”
曙光已现。张女士发现,一种更先进的SMA基因靶向药物已经在国外上市。
如何让更多的罕见病被看见
每一个小群体都不应该被放弃。对于罕见病患者来说,得到早期诊断是一切治疗的前提。但由于病情罕见且确诊困难,很多患者因此错过治疗最佳时机。
为了破解这一难题,帮助更多罕见病群体早诊早治且治疗可及,2019 年,江苏省罕见病诊疗协作网成立。依托协作网,可全面摸底罕见病发病情况,从而为罕见病药物医保政策制定提供依据。
研究显示,罕见病的发病机制与基因、环境、免疫力和内分泌有关,往往有着复杂的临床表现。同一疾病不同时期、不同亚型临床特征不尽相同,甚至变异很大。这不仅增加了罕见病的认识难度,也增加了罕见病治疗的壁垒。
“因此,找到正确的医生、少走弯路更为关键。”事实上,“会看罕见病的医生,比罕见病更罕见。” 江苏省医学会罕见病学分会候任主任委员、江苏省人民医院老年神经内科主任医师牛琦告诉采访人员,部分罕见病有着明显的特征,可快速鉴别。但此类疾病只占罕见病中的极少部分。“更多的罕见病不仅要求医生见识多广、掌握新技术,更多医生的医学影像学、生化检查、如何挑选遗传性检查等专业能力提出要求。有时,为了弄清发病原因、判断是哪一种罕见病,我们不仅仅是只管一个病人,甚至管的是患者一大家子人、好几代人。”

文章插图
如何预防罕见病的发生?张爱华建议,可通过三级预防。一级预防是婚前孕前进行预防性筛查;二级预防是孕期通过基因检测等手段进行罕见病的遗传性筛查;三级预防是新生儿出生后,通过新生儿筛查早诊断,早发现。
对于儿童罕见病的防治,张爱华表示:“有家庭遗传性疾病,或是罕见病家族史的这一类这些人群,我们要尽早地进行相应的筛查,通过基因筛查、遗传咨询防治遗传病。”
诊断困难只是罕见病患者面临的第一个难题,即使能够确诊,大部分患者依然面临无药可用的困境。此外,即使有药可医,大部分罕见病药物价格都极其昂贵,医药费让很多罕见病患者家庭无力承担,只能望药兴叹。
罕见病药物短缺是全世界面临的共同难题。全球大概只有不到10%的罕见病有药可治,罕见病用药面临着患者少、研发周期长、临床数据匮乏、临床入组面临很大挑战、单个市场规模小等诸多困难,这些困难叠加,让许多药企缺乏研发或引进药品的动力。
在牛琦看来,给予药剂研发者充分的条件和时间,提供更多条件支持罕见病药物的研发工作,促进我国原始创新罕见病药物,让更多的罕见病药剂“中国制造”是当务之急。
张爱华表示,让罕见病有药可用仍是全世界面临的共同挑战。“罕见病药物研发依旧任重道远,我们需要各方的协助,共同的努力,来提高我们罕见病药物的可及性。”
此外,也有专家提出,可以建立一个全国统一、公开透明、独立运行的罕见病诊疗及基金信息化的管理平台。“可以利用平台,积极引导患者注册登记,提供必要的诊疗信息,为罕见病患者提供经济补助、生活辅助、信息与资源、服务等方方面面的帮助,慈善机构也可以通过平台去帮助患者,甚至医疗工作者、研究者都可以通过平台去探索新的研究方向,发现新的手段,利用新的技术,更好地服务这个群体。”张爱华说。
新华日报·交汇点采访人员 王甜 叶真
- 其实,罕见病并不罕见|国际罕见病日 | 全人类
- 备孕人群看过来!守好出生缺陷防控 “第一道防线”,这个特别的门诊亮相|国际罕见病日| 妇婴
- 新生|市医疗集团:守护天使 让新生之花健康成长
- 患者|国内患者达2000万,5%的有效治疗方式,谁来“拯救”罕见病患者?
- 人民日报|人民日报关注罕见病:病虽罕见,关爱不能罕至
- 电竞|继GK梦岚回归之后,AG也有人成年重返赛场,同样是让人瞩目的天才
- 遗传|2000多万罕见病患者,90%无药可医
- 罕见肿瘤多学科专家研讨会在沪召开 行业呼吁关注多层次医疗保障体系建立|健康 | 研讨会
- 罕见病|国际罕见病日丨一场跨越20载的生命绽放
- 遗传病|【国际罕见病日】并不“罕见”,应被“看见”