2022年2月28日是第15个“国际罕见病日”,今年的国际主题是“Share Your Colours”(分享你的生命色彩),口号是“Rare is many,Rare is strong,Rare is proud”(罕见并不孤单,罕见即强大,因罕见而骄傲)。

文章插图
根据世界卫生组织的定义,罕见病为患病人数占总人口的0.65‰~1‰之间的疾病或病变,即使按照最低发病率推算,中国的罕见病患者也逾千万。
“人鱼宝宝”(并腿畸形)、“瓷娃娃”(成骨不全症)、“月亮的孩子”(白化病)……这些富有诗意的名字背后,都是罕见病患者难以言说的痛。
尽管目前很多罕见病还不能被完全治愈,但这并不意味着“无计可施”,科学的诊疗手段可以有效控制和缓解疾病症状与发展进程。
自毁容貌综合征:三十八万分之一的痛
2021年,2岁的男孩轩轩(化名)因急性肾功能衰竭由外院转诊至中国科大附一院(安徽省立医院)小儿内科,经过积极治疗,轩轩的感染很快被控制,肾功能也有了明显恢复。
然而,在诊疗中,医务人员发现轩轩存在明显的行为发育异常:快2岁了还不会坐,不会喊妈妈,喂养也很困难,总是哭闹、乱抓。
“轩轩是我的第三个儿子,前两个孩子一个在13个月的时候夭折了,一个在10个月时夭折了,我们家像是中了什么‘魔咒’,孩子总是没办法健康长大。”轩轩的妈妈神情黯淡地告诉医生,几个孩子都存在行为发育落后的情况,前两个孩子还出现过惊厥。
“听轩轩妈妈的描述,我们感觉轩轩很可能得的是一种遗传性疾病。”中国科大附一院小儿内科副主任医师杨春芳说。
通过基因检测,轩轩被确诊为患有一种名为“Lesch-Nyhan Syndrome”的罕见病(简称LNS)。这种病还有个可怕的别名:自毁容貌综合征,发病率只有三十八万分之一。追踪轩轩生后各项检查,杨春芳和小儿内科主治医师朱娟发现轩轩存在泌尿系结石、高尿酸血症,这些都符合LNS的临床表现。按照遗传规律,轩轩的两位哥哥,也很可能是LNS。
“LNS是X染色体连锁隐性遗传性疾病,这类疾病的特点是,在母亲携带致病基因的情况下,生养的男孩一半发病,女孩则一半携带致病基因。”朱娟介绍,由于基因突变引起特定的酶活性缺乏,患者的发育将严重迟缓,出现高尿酸,还会有肌张力障碍和智力障碍。更残酷的是,患者会在成长中出现不受控制地自残行为,如咬嘴唇、咬手指等,即便疼得大哭,也无法停止。这些患者一般生活质量很差,生命很短暂。
明确诊断后,经科室讨论,给予了轩轩低嘌呤饮食,别嘌醇降尿酸及碱化尿液、多饮水的治疗方案,并建议配合康复训练。经过努力,轩轩尿酸长时间控制在正常范围。“轩轩吃饭乖了,会笑了,也能牵着走了。”前不久复诊时,妈妈欣慰地告诉医生。
朱娟提醒,尽管目前对于LNS还没有根治的手段,但如果家长发现孩子发育迟缓,有认知与行为障碍,应尽快到专业医疗机构儿科就诊,尽早发现及诊断疾病,控制高尿酸血症,对患儿及家庭生活质量将有很大改善,致病基因的明确也会为患儿家庭再生育提供遗传咨询。
中国科大附一院小儿内科一直致力于罕见病诊治,通过线上线下相结合的方式,持续关注罕见病患儿疾病的发展,改善患者及其家庭的生活质量。近年来诊断罕见病包括:非经典性溶血性尿毒症、自身免疫性脑病、结节性硬化、全身型重症肌无力、进行性脊髓性肌萎缩、家族性地中海热、法布雷病、糖原累积症(I、II型)、瓜氨酸血症、原发肉碱缺乏症、家族性高胆固醇血症、小胖威廉综合征等。尤其血液净化术的应用挽救了多名患者的生命。
17α-羟化酶缺陷症:“另类”女孩的隐忧
17岁花季少女小婉(化名)的妈妈,在中国科大附一院内分泌科门诊诊室里满面愁容,原因是女儿都这么大了竟然一直没有来过月经。小婉和医生说,不少小伙伴在小学六年级、初一就已经来“大姨妈”了,而她却一直没有来,而且胸部也没有任何变化,在学校里感到自己很“另类”。妈妈说,带小婉去了很多医院看病,都没查出来病因。
医生为小婉进行了一系列检查。影像检查很快有了发现,CT片上显示小婉双侧肾上腺明显增粗,即使小婉并没有高血压、低血钾之类症状,但是根据科室之前的相关研究结果,一些先天性肾上腺增生患者为部分性联合缺陷,可以只表现为原发性闭经,因此医生为小婉进行了基因检测。
基因检测证实了小婉患有一种罕见的内分泌疾病——17α-羟化酶缺陷症。
内分泌科主任叶山东介绍,17α-羟化酶缺陷症是先天性肾上腺皮质增生的一种少见类型,为CYP17A1基因突变引起的常染色体隐性遗传病,发病率在五万分之一至十万分之一。这种疾病主要表现为高血压、低钾血症,女性表现为性幼稚、原发性闭经,男性可有假两性畸形。
“谜底”终于水落石出,医生帮助小婉通过使用性激素替代疗法,促进第二性征的发育以及获得正常的骨代谢,并通过雌激素联合孕激素治疗诱导人工月经周期。一直压在小婉心里的石头终于落了地。
近年来,中国科大附一院内分泌科在罕见病临床与基础研究方面取得了一些成果:在国际上首次报道了糖原累积症Ia型患者G6PC基因的新错义突变,并被人类基因突变数据库(HGMD)收录;首次报道了假性甲状旁腺功能减退症合并Turner综合征并初步阐明了致病机理;在国内首次报道了成年期Ia型糖原累积症致继发性骨质疏松症的家系。此外在Alstrom综合征、Bardet-Biedl综合征、DiGeorge综合征等内分泌罕见病方面也提出了新见解,为很多内分泌系统罕见病患者明确了诊断和治疗方案。
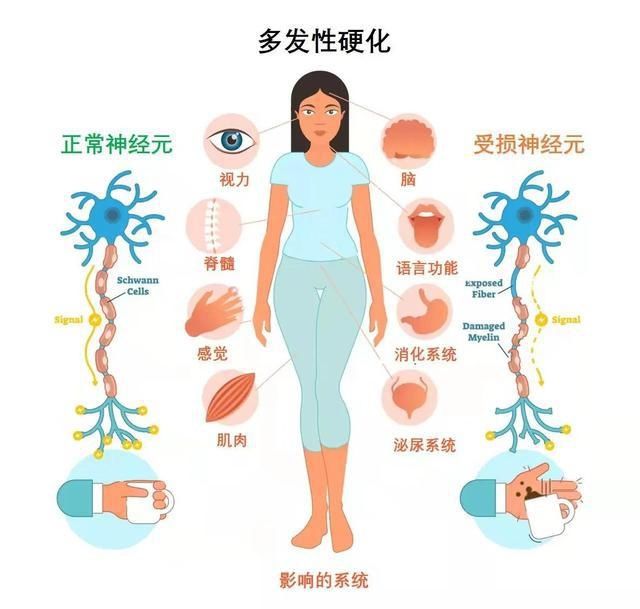
多发性硬化:身患“美女病”的花季少女
2021年6月,正在读高二的17岁女孩小美(化名)发现自己视力越来越模糊,家长以为是近视了,带小美去了当地医院眼科,想着配个眼镜也许就能解决问题。但眼科医生接诊后发现事情没有这么简单,小美的视神经有受损的情况,建议他们去神经内科看看。
更糟的是,很快,小美发现自己走不稳路,讲话也不太灵活,在当地医院做了头颅和脊髓磁共振后,诊断为“视神经脊髓炎”。在激素冲击治疗后症状好转,但是好景不长,1个多月之后,小美再次出现行走不稳,头晕、看东西重影的症状。
辗转多家医院后,2021年9月,家人带小美来到中国科大附一院神经内科就诊。副主任医师江艳和徐文根据小美症状及病史为她进行了腰椎穿刺、自身免疫脱髓鞘抗体检测、磁共振复查、并组织了多次治疗组病例讨论,最终小美被确诊为“多发性硬化(复发-缓解型)”。
江艳介绍,多发性硬化(MS)是一种中枢神经系统慢性炎性脱髓鞘性疾病,好发于20-40岁的中青年,是目前全球最常见的青壮年致残性神经系统疾病之一。在亚裔人群的患病率仅为五万分之一至十万分之一。女性的患病率约是男性的2倍,因此这种疾病又被称为“美女病”。
文章插图
“肢体运动障碍、感觉障碍、疲劳和平衡障碍等都是多发性硬化常见的临床表现。85%多发性硬化属于像小美一样的复发缓解型,作为一种病程进展性疾病,如果未能得到有效治疗,约50-60%的患者最快5年会进展为继发进展型多发性硬化。患者可能出现无法缓解的行走困难、认知损害、视力损害等症状,严重影响工作和生活。”江艳说。
经科室讨论,医生为小美制定了免疫修饰(DMT)治疗方案,使病情得到了有效控制,症状也得到了缓解。“最近都没有发现身体有什么异常,现在和生病前一样,可以自由自在的活动,现在我正在努力备战高考,希望能考到理想的大学。”复诊时,小美高兴地对医生说。
徐文表示:“神经免疫领域的很多疾病都属于罕见病,除了多发性硬化(MS),还有视神经脊髓炎谱系疾病(NMOSD)、MOG相关脑病(MOGAD)、重症肌无力(MG)等等,这类疾病发病率低、诊断困难、治疗困难,易反复复发。对于这类疾病的治疗,我们除了常见的免疫抑制剂、激素治疗之外,还会根据患者病情采取免疫修饰(DMT)治疗、单克隆抗体、蛋白A免疫吸附治疗等最新的治疗技术,可明显改善患者症状及预后。”
中国科大附一院神经内科在诊治神经免疫罕见病方面具有丰富的经验,在科主任王国平教授的带领下,将“神经免疫疾病诊治”作为科室亚专科重点方向加强扶持和建设。学科参与了多项全国多中心的神经免疫病的临床药物试验及研究,均取得可喜进展。
学科神经免疫团队还针对多发性硬化、视神经脊髓炎、重症肌无力等疾病专门定期开展患教学习班,建立了相关疾病的专属管理群,便于加强患者管理、规范治疗和医患沟通。
罕见病不“罕见” 积极预防是关键
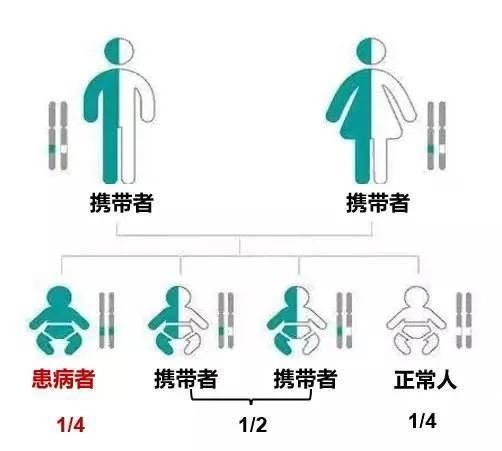
罕见病好像离我们很遥远,但又真实地出现在我们身边。中国科大附一院妇产科产前诊断中心亚专科副主任吴丽敏介绍,罕见病中70-80%是基因遗传病,超过84%的人至少携带一个隐性遗传病的致病变异,平均每人携带3-5个隐性遗传病致病突变。如果夫妇俩同时携带同一个基因的致病突变,那么在毫无干预的情况下,每一次生育都像掷骰子,子代呈一定概率患病。
文章插图
▲常染色体隐性遗传疾病的遗传模式
吴丽敏提醒,把好预防关,早发现、早干预是罕见病防治的最佳途径。通过婚前、孕前筛查,产前筛查和产前诊断,新生儿筛查的三级预防策略,可以有效防范罕见病的发生。
一级预防——孕前。加强婚检、孕前进行优生遗传咨询、高危人群给与遗传病携带者筛查,全面排查从源头上予以预防。
二级预防——产前。通过孕期产检可以筛查出高危孕产妇,及时予以产前诊断和筛查,阻断罕见病先天缺陷患儿出生。
三级预防——新生儿。部分罕见病,如一些遗传代谢疾病、内分泌异常疾病等,通过新生儿疾病筛查能及时发现,尽早治疗,伤害可降到最低。
【 小儿内科|罕见病不“罕见”!我们该如何预防?】(安徽城市之声:蔡佳 通讯员:朱娟 祝捷 徐文 宗璐 方雯)
- 协作组|广东省溶酶体贮积症诊疗协作组成立 打造罕见病诊疗“样本”
- 湖南省|湖南启动罕见病诊疗协作网线上平台
- 识别|提升罕见病识别意识和诊疗水平
- 疾病|关注罕见病 更要重视疾病后续管理及治疗
- 张抒扬|国际罕见病日:从“病无医”到“早确诊”
- 亚硝酸盐|91岁院士,我国肿瘤内科学创始人之一,提醒少吃3物多做5事
- 明天会更好|关注罕见病:江苏成立援助专项基金 推动持续有效治疗
- 北京大学|北大三院肿瘤内科医生张煜被解聘,院方:对医院声誉造成不良影响
- 治疗|关注罕见病:破解“天价药”困境
- 患者|罕见病患者在北京协和医院平均确诊时间不到4周