百度计算生物研究登Nature子刊!结果超斯坦福MIT,落地制药领域
杨净 萧箫 发自 凹非寺
量子位 | 公众号 QbitAI
百度新研究,登上了Nature子刊。
科技公司卷到学术圈顶刊上不算稀奇。
但这次有点不同寻常。
研究领域与生物领域直接相关,接收该论文的期刊Nature Machine Intelligence(NMI),影响因子达到了16.649。

文章插图
除了专业度保障,研究的实验结果也超越MIT斯坦福。
而且更关键的在于,跟后者大部分“产学研”模式不同。
百度是实打实自己独立搞出来的——
作者全部来自螺旋桨PaddleHelix,百度生物计算团队。
嗯,还是可复现的那种,目前GitHub上已经开源了完整代码(地址可在文末获取)。
研究人员表示,相关部分项目已经实现了商业化落地。
来看看究竟是一项什么样的研究。
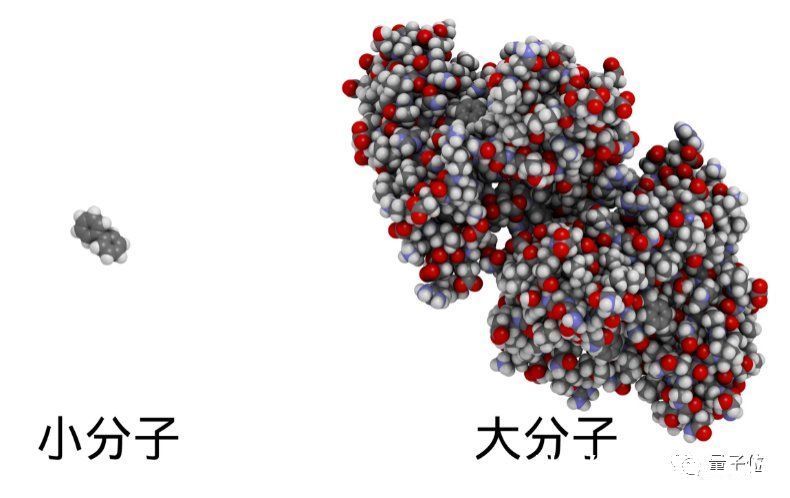
小分子3D结构被AI整明白了此次百度聚焦的研究,是小分子化合物性质预测。
简单来说,通过小分子结构来预测其性质,帮助药物研发的早期探索,从而解决该领域成本高、时间长、成功率低等难题。
小分子药物结构有良好的空间分散性,其化学性质也更有助于成药,因此相较于大分子药物(蛋白质、核酸等)在药物研发上更有优势。市场上大部分药物也属于小分子药物。
文章插图
但即便有先天优势,面临的特殊挑战也不小。
最大的挑战,莫过于小分子的筛选空间实在是太大了。
早前Nature一篇研究表明,小分子药物研发筛选数量在10的60次方。
什么概念呢?作者形容,“比太阳系的原子数量还要多”。
要在这样一个庞大「小分子宇宙」中寻求合适的候选药物,高效准确的化合物表征就起到关键作用。
基于这样的背景下,研究团队此次的研究提出了几何增强型的分子表征方法,简称GEM。
这个方法主要包含两个部分:基于空间结构的图神经网络GNN、以及多个几何级别的自监督学习。
不难看出,本次研究的亮点在于空间、几何。
据介绍,这是业界首次将空间结构引入到化合物建模当中。
之所以这样强调,跟他们要解决的问题不无关系,那就是让AI也能理解小分子的3D结构。
个中原因,需要从现有表征方式说起。
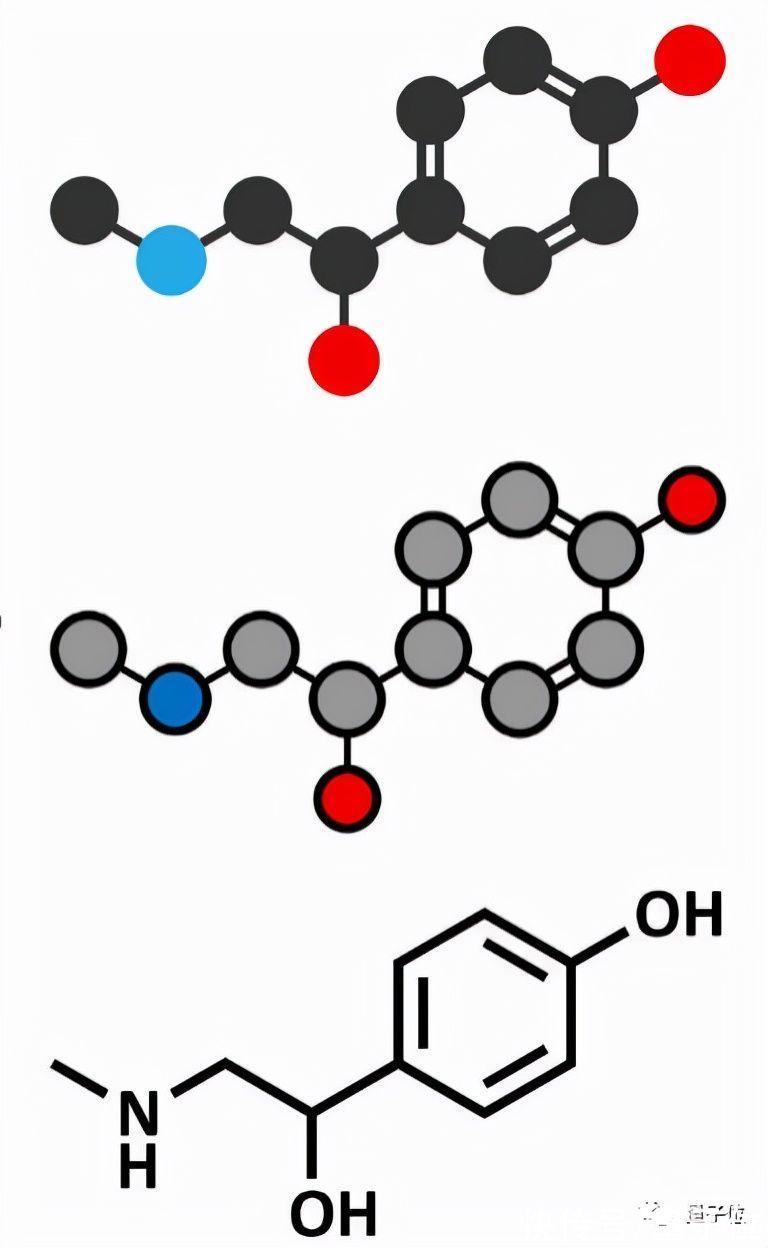
目前研究主要有两种表征方式:基于序列的一维表征和基于图形的表征。
一个以字符串作为输入,利用序列模型比如RNN和Transformer来学习分子表征,但存在一些明显的局限性,比如字符串语法难以理解,两个相邻的原子在文本序列上可能相距甚远;字符串的一个小变化可能导致分子结构的大变化。
另一个则与今天的研究相关——GNN建模,以图作为输入,每个原子是一个节点,每个化学键是一个边。
嗯,就跟化学书那样式儿的。
文章插图
但多数研究只停留分子的二维信息,忽略了三维空间结构。
这也不难理解,毕竟要想准确获得分子的三维结构信息其实并不容易。
要是所选描述三维结构的参数一旦不理想,其性能可能上述两种表征方法更糟,还将面临鲁棒性不足和预测性能不理想等问题。
但即便如此,三维结构信息却很关键,因为往往决定了分子的物理化学性质及生物活性的不同。
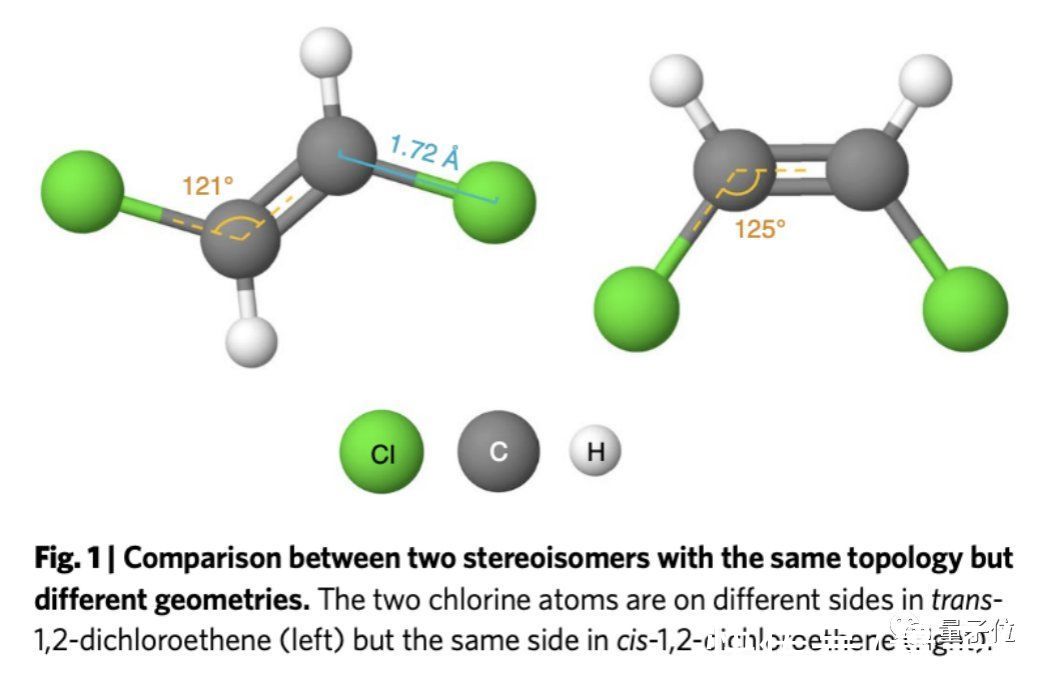
最典型的例子,就是高中学的同分异构体。
以二氯乙烯为例,它就有顺反式结构,因为几何结构不同,导致二者的水溶性不同。
文章插图
还有像顺铂和反铂(二氯二氨合铂),顺铂是一种流行的抗癌药物;但反铂有毒却没有抗癌活性。
既然如此,那就来看看这项研究是如何解决的。
首先来看图神经网络,本次研究人员提出了一种GeoGNN。每个分子的输入包含两个图,可同时模拟原子、键和键角的影响。
第一个图,即二维结构图,也叫做原子-化学键图,仍以原子为节点,键为边。
第二个图,化学键-键角图,则是以键视作节点,键角视作边。
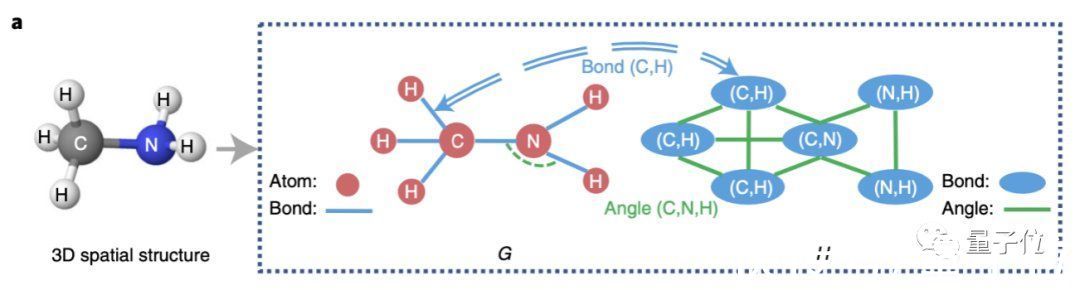
文章插图
GeoGNN经过多轮迭代学习原子和键的表征向量,为了连接两个图,化学键作为每一轮迭代中图G和图H的桥梁进行信息互通。
最后通过汇集原子表征得到分子表征,用来化合物性质预测。
为了更好的学习分子空间知识,除了以几何信息作为输入,进一步地,研究团队设计了多项自监督学习任务。
- 百度|英特尔计划500元销售400万块Arc显卡,性能比锐龙核显还慢
- 资本市场|“东数西算”下中国云计算仍在蓝海
- 药物|“自降身价”也要上市,乐普生物离盈利之路有多远?
- 百度|游戏手机渐入佳境,ROG这款老旗舰配置拉满,现在入手依旧能打
- 电池|电池为信息保存提供电源,如果没有电池,计算机中就没有时间概念
- neuron|38岁青年生物学者周传去世,饶毅悼念“指导过老师的学生”
- npu|影像力表现最大化,OPPO Find X5系列将实现计算摄影新突破
- 云计算|东数西算+云计算+大数据, 算力已覆盖36个城市, 为华为打造数据中心
- 智慧医疗|百度、科大讯飞抢食智慧医疗奶酪
- 我们和计算生物学从业者深度聊了聊|对撞派 · 圆桌实录