npj: 参数变力为能量—加速机器学习势函数构建
 文章插图
文章插图
基于第一性原理计算开展机器学习来训练经验势函数是近年材料计算领域最重要的进展之一 。 精确训练的势函数具有可以比拟第一性原理的精度 , 但计算量显著降低 , 其计算量仅与模拟原子数成线性关系 , 而典型的密度泛函计算一般与原子数成三次方关系 。 因此 , 基于机器学习经验势可以用来高精度地模拟超大规模的复杂材料体系 。 准确的机器学习经验势需要同时利用第一原理获得的力和能量来同时训练 。 由于力是能量的一阶导数 , 这导致需要十分庞大的数据集才能实现准确的训练结果 , 同时对训练方法也提出很大挑战 。
来自美国、德国和英国的联合研究团队 , 提出将能量用原子间作用力泰勒展开 , 因此巧妙地绕开了上述问题 。 基于泰勒展开的能量表达式 , 他们提出了新的势函数训练方法 。 计算结果表明 , 相比直接采用作用力训练势函数的方法 , 该方法不仅将势函数的精度提升了50% , 同时计算效率明显提升 。 为进一步验证该方法的有效性 , 作者选取了三个具体的模型体系 , 即水分子团簇、液态水和复杂金属氧化物开展势函数训练 。 他们发现 , 该方法可以显著降低训练所需的数据集大小;具有很好的可移植性 , 可以准确预测数据集之外的新体系;同时可以提升作用力预测的准确性 。 简而言之 , 该方法简化了神经网络经验势的构造过程 , 有望推广应用于任意类型的材料中 。
【npj: 参数变力为能量—加速机器学习势函数构建】该文近期发表于npj Computational Materials 6: 54 (2020) , 英文标题与摘要如下 , 点击可以自由获取论文PDF 。
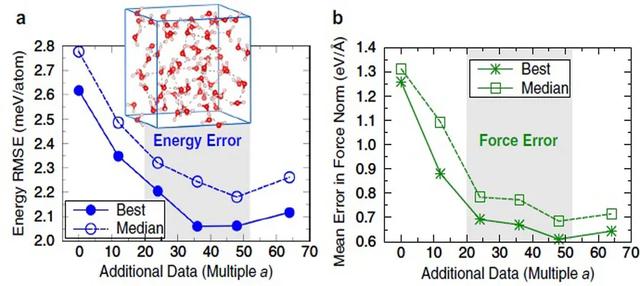 文章插图
文章插图
Efficient training of ANN potentials by including atomic forces via Taylor expansion and application to water and a transition-metal oxide
April M. Cooper, Johannes K?stner, Alexander Urban & Nongnuch Artrith
Artificial neural network (ANN) potentials enable the efficient large-scale atomistic modeling of complex materials with near first-principles accuracy. For molecular dynamics simulations, accurate energies and interatomic forces are a prerequisite, but training ANN potentials simultaneously on energies and forces from electronic structure calculations is computationally demanding. Here, we introduce an efficient alternative method for the training of ANN potentials on energy and force information, based on an extrapolation of the total energy via a Taylor expansion. By translating the force information to approximate energies, the quadratic scaling with the number of atoms exhibited by conventional force-training methods can be avoided, which enables the training on reference datasets containing complex atomic structures. We demonstrate for different materials systems, clusters of water molecules, bulk liquid water, and a lithium transition-metal oxide that the proposed force-training approach provides substantial improvements over schemes that train on energies only. Including force information for training reduces the size of the reference datasets required for ANN potential construction, increases the transferability of the potential, and generally improves the force prediction accuracy. For a set of water clusters, the Taylor-expansion approach achieves around 50% of the force error improvement compared to the explicit training on all force components, at a much smaller computational cost. The alternative force-training approach thus simplifies the construction of general ANN potentials for the prediction of accurate energies and interatomic forces for diverse types of materials, as demonstrated here for water and a transition-metal oxide.
- 充电宝到底该怎么选呢?重点看这五个参数放心选
- realmeRace和realmex7pro哪个好值得入手 参数对比评测
- Linux中MySQL配置文件my.cnf参数优化
- 品牌|摩托罗拉G10手机外观和参数曝光!上市时间还未明确
- 骁龙888和三星Exynos2100哪个好 性能参数对比评测
- 华为mate|OPPO FindX3参数确认,硬件达到天花板,3K屏成亮点
- 天猫|骁龙750g和765g哪个好 750g和骁龙765g区别参数对比
- IT之家|4600U 一致,AMD R5 5500U 现身:6 核 12 线程,参数与
- 快手视频|三星2100和1080哪个好,性能跑分参数全对比评测
- 一加科技|一加9Pro手机参数出炉:骁龙875+3K屏!